Review Article
Cellular Senescence and their Role in Liver Metabolism in Health and Disease: Overview and Future Directions
Schade M1, Sanabria JA1, Aguilar R1, Modarresi M1, Gillon B1, Hunter Z1, Fannin J1, Mallick A1, Brunengraber H2 and Sanabria J1,2*
1Department of Surgery, Marshall University, USA
2Department of Nutrition Proteomic and Metabolomic Core Facility, Case Western Reserve University, USA
*Corresponding author: Kjell K Ovrebo, Department of Surgery, Haukeland University Hospital, 5021 Bergen, Norway
Published: 13 Jul, 2018
Cite this article as: Ovrebo K, Berget E, Grong K, Fevang
J. Strangulation Obstruction and the
Release of Strangulation. Effects of
Fluid Administration on Mucosal Blood
Flow and Damage. World J Surg
Surgical Res. 2018; 1: 1031.
Abstract
Chronic liver disease has globally risen mainly due to a prevalent Hepatitis C Virus (HCV) infection
rate and an epidemic of obesity. It is estimated by the year 2030, 2.2 billion people around the
world will be overweight and 1.1 billion people will be obese. Diabetes and obesity are the main risk
factors for the development of the metabolic syndrome and in the liver of Non-Alcoholic Fatty Liver
Disease (NAFLD) which could progress to NASH related cirrhosis and liver malignancy. At present
there is not effective therapy for NASH besides loss of weight and exercise. Furthermore, optimal
management of HCC with curative intent includes resection or liver transplantation. Therapies
that is limited due to the degree of liver dysfunction, medical conditions at the time of diagnosis
and the scarcity of available liver grafts. The role of cellular lipid management and metabolism
in human health and disease is taking a center stage. The present overview articulates the current
pathophysiology of fatty liver disease under the aging processes, potential biological markers of liver
disease diagnosis and progression and future therapies.
Keywords: Review; Senescence; Cell aging; Mitochondrial function; Cancer; Obesity; NASH;
ROS
Abbreviations
ADP: Adenosine Diphosphate; AGPAT: sn-1-acyl-glycerol-3-phosphatase Acyltransferase; ATP: Adenosine Triphosphate; CACT: Carnitine-Acylcarnitine Transferase; DGAT: sn-1,2- diacylglycerol Acyltransferase; DBC-1: Delete in Breast Cancer-1; DM: Diabetes Mellitus; ECM: Extracellular Matrix; ER: Endoplasmic Reticulum; FA: Fatty Acids; Fox01: Fork Head Box Protein; GBD: Global Burden of Disease; GPAT: Glycerol-3-phosphatase Acetyltransferases; GSH: Reduced Glutathione; GSK-3: Glycogen Synthase Kinase 3; HBV: Hepatitis B Virus Infection; HCV: Hepatitis C Virus Infection; HSC: Hepatic Stellate Cells; HSP70: Heat Shock Proteins; HTN: Hypertension; IL-1ß: Interleukin-1 Beta; IL-6: Interleuking-6; INF-α: Interferon Alpha; JNK’s: c-Jan NH2-terminal Kinases; LD: Lipid Droplet; MAPK: Ras-MAP Kinase; MF: Portal Myofibroblast; MMP: Matrix Metalloproteinases; MPTP: Mitochondrial Permeability Transition Pore; NAFLD: Non-alcoholic Fatty Liver Disease; NASH: Non-alcoholic Fatty Steatohepatitis; NF-κB: Nuclear Factor κB; NO: Nitric Oxide; PAP: Phosphatidic Acid Phosphatase; PI3K: Phosphatidylinositol 3-kinase; PKC: Protein Kinase C; SMase: Neural Smase; ROS: Radical Oxygen Species; SA- ß-GAL: ß-galactosidase; SAH: Senescence Associated Heterochromatic Foci; SASP: Senescence Associated Secretary Phenotype; SEC: Sinusoidal Endothelial Cells; SGK: Serum/glucocorticoid Kinase; TAG: Triacylglycerol; TCA: Tricarboxylic Cycle; TNF-α: Tumor Necrosis Alpha; TOR: Target of Rapamycin; TIMP: Tissue Inhibitor Of Metalloproteinases; UPR: Unfolded Proteins Response; 4RBP: Factor 4E Binding Protein
Introduction
Global burden of chronic liver disease
Chronic liver disease has globally risen mainly due to a prevalent Hepatitis C Virus (HCV)
infection rate and an epidemic of obesity [1-7]. Between 1990 and 2013, global viral hepatitis deaths
increased from 0.89 million to 1.45 million and in 2013, viral hepatitis was the seventh leading cause
of death worldwide [7]. While HBV infection is decreasing in most endemic areas due to successful
vaccination policies, HCV infection lacks the benefits of a vaccine [7]. Even though HVC antiviral
therapies recently introduced in clinical practice are highly successful, its implementation is limited
due to access and/or financial constraints. Morphological studies in
HCV showed hepatocyte lipid accumulation similar to the one that
occurs in obesity.
It is estimated by the year 2030, 2.2 billion people around the
world will be overweight and 1.1 billion people will be obese [8,9]. In
addition, 36.1% of adult men and 32.4% of adult women had metabolic
syndrome in 2010 [10]. Obesity represents the core component of
the metabolic syndrome, a cluster of metabolic disarrangements
including dyslipidemias, insulin resistance status, hypertension and
organ metabolic disturbances such as Non-Alcoholic Fatty Liver
Disease (NAFLD) and its inflammatory component NASH, diabetes,
nephropathy, cardiomyopathy and muscle dysfunction [10].
Hepatocellular Carcinoma (HCC) has been reported more often in
non-cirrhotic livers in the background of NASH and its risks factors
include male gender, older age, cigarette smoking, obesity and insulinresistant
states [3,4,11]. Overweight and obesity was associated with
an overall increase in liver cancers of 17% and 89%, respectively
[2,11,12] and males with a BMI>35 had a 3.5 to 4 times increase in liver
cancer [12] (Figure 1). Optimal management of HCC with curative
intent includes resection or liver transplantation. Therapies that is
limited due to the degree of liver dysfunction, medical conditions at
the time of diagnosis and the scarcity of available liver grafts. The role
of cellular lipid management and metabolism in human health and
disease is taking a center stage [13]. Higher fat consumption, decline
in physical activity and a progressively aging population are among
the social and behavioral roots of this phenomenon that add to the
genetic load [1]. An overview of the role of cell aging and senescence
in liver metabolic responses to high caloric intake will be performed
in the pages to follow.
Regeneration, necrosis, apoptosis and senescent: a
constant changing balance
The liver is a unique organ with an innate ability to regenerate
through mass compensation to satisfy portal flow and metabolic
demands [14]. After injury and cell necrosis, immune recall of resting
cells occurs and activation of oval-precursor cells in conjunction with
platelets migration switches to a cell division renewal cycle [15,16].
Mitosis is more prominent at the peri-portal stem cell niche site (zone
1) assuring clonal expansion until reaching zone 3 (peri-central vein)
[15,16]. In health, liver mass homeostasis is closely regulated through
a delicate balance among regeneration, apoptosis (programmed cell
termination) and senescence. During states of acute liver injury, the
pendulum moves towards a regenerative and repair phase, however,
during chronic states of liver injury collagen synthesis and deposition
persists leading to organ fibrosis. In addition, natural processes of
organ aging play a main role in organ response to both acute and
chronic injuries. Primary cell life span is determined by a limited
number of cell duplications, the so called Hayflick limit [17]. After
such limited divisions, cells enter a state of cell replicative senescence
which is believed to be triggered by shortening of telomere ends.
Replicative cellular senescence is a stable form of cell arrest
characterized by a lack of cell proliferation activity and apoptosis
resistance mediated through a lack of mitogen response even though
the cells remain metabolically active. On the other hand, cells can be
induced to a senescence status by a variety of cellular stressors such
as DNA damage, UV light, radiation, oncogene activation, increased
H2O2 production and heat stress [17,18]. Senescent cells undergo
morphological changes as they acquire an enlarged and flattened
morphology, in addition to an increase expression of the Senescence
Associated markers ß-Galactosidase (SA-ß-GAL), an accumulation
of the Senescence Associated Heterochromatic Foci (SAHF) and
DNA damage foci, and the expression of the Senescence Associated
Secretary Phenotype (SASP) [18]. Senescent status is achieved and
maintained by active signaling of p53, a tumor suppressor gene that
exercises its effects through activation of p21, a potent cell cycle
inhibitor, and the p16-retinoblastoma protein [18]. Cells induced into
an irreversible cell cycle arrest at the G1 phase will undergo metabolic
disturbances with an increase Reactive Oxygen Species (ROS)
production, decrease Adenosine Triphosphate (ATP) synthesis and
accumulation of lipofuscin [17].
Changes in the content of daily oral intake can influence life span
and thus cell aging. The cell death-inducing DNA fragmentation
factor α-subunit-like effector A (Cidea) is a transcriptional coactivator
implicated in lipid accumulation, cell stress and cell aging. Authors
showed in rodents, that a high lipid diet up-regulated Cidea with
hepatic lipid accumulation, cell stress, mitochondrial dysfunction
and genetic upregulation of aging [19]. Other studies, in support
of these findings have shown a life span reduction up to 30% in
genetically obese mice (ob/ob) and this reduction were reversed by
a caloric restricted diet [19]. Lipid enriched diets are associated in
humans with DM type 2, HTN and cardiovascular events all of which
limit life span. Caloric restriction without malnutrition can extend
life span while caloric excess has the opposite effect [20]. Thus, the
choice of oral intake has a profound impact on life span.
The free radical hypothesis of cell aging still remains the most
reasonable in the induction and maintenance of the senescent status
[17,21]. ROS, reactive nitrogen sp., and lipid peroxide are important
regulators of cell signaling that provides reliable maintenance of
cellular components, support redox-state and regulate the function
of highly metabolic active cells as in hepatocytes and immune cells
[20,21]. ROS in excess from over-oxidation of lipids, proteins, nucleic
acids and other macromolecules is associated with a violation of their
functional activity, reactions that if they last through the cell cycle
can lead to permanent cell dysfunction and/or accelerated aging
cell process. Thus, an excess of food intake in form of continuous
lipid charge will test the oxi-redox systems that keeps the fragile
mitochondrial equilibrium in balance. Continuous metabolic
stress changes the equilibrium towards lower levels of antioxidants
(glutathione sp.) with further increase of ROS accelerating processes
of apoptosis and senescence. Former processes in turn lead to arrest
of regeneration and activation of Hepatic Stellate Cells (HSC) and
therefore fibrogenesis (Figure 2A).
Hormonal signaling modulates cellular response to caloric
intake. Insulin and somatotropic signaling are critically important
not only in the control of aging and longevity under conditions of
unlimited food supply but also in mediating the effects of caloric
restriction on life span. In a rodent model of thyroxine induced aging,
thermogenesis was directly correlated with increased mitochondrial
function, increased ROS production, decreased concentration
of reduced glutathione, reduction in the activity of antioxidants
enzymes and increased senescent marker expression in the liver as
well as in other organs [21]. Estrogen influences lipid metabolism
through nuclear receptors which enhances apoptosis of mutated
cells, improves mitochondrial function, and decreases the metabolic
syndrome phenotype [10,22]. Actions this may explain, at least in
part the constant disparity of overall life expectancy by gender.
Lifestyle changes such as exercise and caffeine supplementation
have shown to increase the ratio of reduced/oxidized glutathione
liver and muscle tissue in the rodent model [23]. Although liver
enzymes were identical in experimental and controls groups, plasma
levels of cytokines associated with inflammation (IL-1ß, IL-6, TNF-α
and INF-α) and cell aging were found to be significantly decreased in
the experimental group when compared to controls [23]. It was noted
that although exercise increased the production of ROS, exercise also
evoked a beneficial increase in levels of cell antioxidants, and lowered
levels of oxidative damage when cells were exposed to a second
injury, i.e. lipid charge. Thus, the concept of exercise inducing gene
expression of antioxidant enzymes that may protect the cell from
other insults was called ‘hormesis’ [23]. Although caffeine, a member
of the methyl-xanthine family increased the ratio of reduced-oxidized
glutathione, no other markers of cell stress were modified. Perhaps,
caffeine potentiates further the beneficial effects of exercise.
Liver metabolism in health and disease
The reduced tri-peptide Glutathione (GSH) is the major
antioxidant in the body responsible for maintaining the intracellular
redox balance. GSH in plasma represents 90% of the GSH synthesized
in the liver [24] and aging is associated with a progressive decline in
the levels of GSH in humans and rodents [25]. Senescent liver cells in
culture showed elevated ROS leading to a state of chronic oxidative
stress. In addition, age associated decline in GSH has been linked to
an activation of neural sphingolipid hydrolase enzyme (NSMase) and
the accumulation of bioactive ceramide, a precursor of inflammation
[25]. The availability of L-cysteine is the rate-limiting factor of
GSH synthesis and oral supplementation of cysteine alleviates GSH
deficiencies in humans and rodents [25]. GSH deficiency can be
alleviated by the oral intake of cysteine and its restoration rates appear
to be age and sex dependent. Older animal models are associated with
increased cellular stress and an enhanced subcellular injury after heat
stress associated with an increased iron intracellular deposition [26].
These cause damages to mitochondria and lysosomes. Although a
more precise mechanism of organelle damage was not enunciated,
iron deposition mediated decrease in transferrin-receptor-1 which
upregulates the iron storage protein ferritin after heat stress.
Nevertheless, the synthesis of the iron exporter protein ferroportin
was delayed [26]. Effect this may explain at least in part, organelle
damage in the aging cell that occurs after natural oxidants depletion
(Figure 2B).
A diet enriched in calories and lipids increases free Fatty
Acids (FA) in plasma obligating cells to protect themselves from
lipotoxicity or death by either oxidizing FA’s or sequestering them as
Triacylglycerol (TAG) within Lipid Droplets (LD) [13]. An exerciseinduced
transcriptional coactivator PGC-1α appears to play a key
role in coordinating intramuscular LD-signaling with mitochondrial
remodeling. TAG within lipid droplets are the major form of energy
storage in the body (muscle, liver, fat tissue) and a reservoir of
membrane lipid component. TAG synthesis is initiated by Glycerol-
3-Phosphatase Acetyltransferases (GPAT) at the mitochondrial
and sarcoplasmic reticulum membranes and it is completed at the
sarcoplasmic reticulum by the sn-1-Acyl-Glycerol-3-Phosphatase
Acyltransferase (AGPAT), Phosphatidic Acid Phosphatase (PAP)
and sn-1, 2-Diacylglycerol Acyltransferase (DGAT) [13]. Synthetized
LD-TAG is localized preferentially in proximity to mitochondrial
membranes named “contact zones”. Once TAG’s are released, they
are mainly used in the mitochondria for ATP synthesis via oxidative
phosphorylation from the ß-oxidation path. The “athlete paradox”
states that the accumulation of TAG in the trained and insulin
sensitive cells is in greater proportion than the TAG accumulation in
cells from diabetic subjects with insulin resistance. This observation
supports the hypothesis of mitochondrial dysfunction as a factor of
TAG accumulation from a sustained lipid charge.
The protein family of Perilipins (PLIN) is associated with LD’s
and their scaffolding may affect the interaction between TAG and the
mitochondria [13]. The PLIN family consists of PLIN1 to 5; the most
common PAT (perilipin/ADRP/TIP47) interacts with LD in different
proportions. In the liver, down-regulation of PLIN2 promotes a
reduction of hepatic steatosis and increases insulin sensitivity, albeit
a reduction in both PLIN2 and PLIN3 cause insulin resistance [13].
In the heart, a PLIN5 deficiency causes increased lipid oxidation,
increased ROS production and decreased cardiac function. In heart
and skeletal muscle TAG and FA are the main metabolic source of
energy through the ß-oxidation pathway, suggesting a very tightly
regulated process from cell storage to mitochondrial metabolic use.
While TAG may come from LD, FA’s are mainly transported in plasma
as albumin-bound or as part of the Very Low Density Lipid-protein
(VLDL) complex. Different transmembrane transporter systems are
involved in their translocation to the inner cell compartment where
the Long Chain Fatty Acid (LCFA) forms thioesters with coenzyme A
(CoA). LCFA-CoA can form TAG for storage as LD, or can enter the
outer mitochondrial membrane where CPT1 catalyzes the reaction of
LCFA-CoA to LC-acylcarnitine. The former compound can actively
cross the inner mitochondrial membrane with the exchange of
carnitine for acylcarnitine. CACT is highly expressed in tissues with
predominant ß-oxidation metabolism.
The metabolism of FA in the mitochondrial matrix is sequentially
catalyzed through a ß-oxidation process by four enzyme families:
acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-hydroxyacyl-
CoA dehydrogenase and 3-ketoacyl-CoA thyolase. While acylcarnitine
is converted back to acyl-CoA to enter the TCA cycle,
dehydrogenases activity shows different affinity for short, medium,
long and very long FA‘s [13]. Every cycle of ß-oxidation renders
acyl-CoA and shortens the FA chain by 2 carbons providing the
equivalents of electron donors NADH and FADH2 which is the
driving force for the synthesis of ATP. Although the ß-oxidation
pathway is an effective way of ATP production, an overload of FA
may play the role of uncoupler that exercises an inhibitory effect on
the respiratory chain through a proton-phoric effect on the inner
mitochondrial membrane. This effect results from the implicit effect
of FA on the mitochondrial membrane permeability by opening of
the permeability transition pore, which results in the subsequent loss
of electrical gradient and arrest of the respiratory chain [13]. The
above concept favors the hypotheses of mitochondrial dysfunction
from FA overload as the primary step of the insulin resistance state
in obese patients. Other authors have found no mitochondrial
respiratory changes in the steatotic liver but in mitochondria from
skeletal muscle from a rodent model of high fat diet induced obesity
[27]. Authors have proposed mitochondrial changes are due to an
adaptation of the mitochondria to the high lipid charge rather than
a defect per se in its function. No explanation was provided by the
authors regarding the increase in ROS and inflammatory changes
observed. Others suggested a protected effect from caloric lipid
surplus against the development of metabolic dysfunction, as long
as cells maintained functional adipocyte storage with low levels of
tissue inflammation [28]. Once adipose storage capacity is saturated,
fatty excess spills over into other tissues leading to LD accumulation
with subsequent lipid oxidation and an inflammatory response which
precedes the metabolic syndrome manifestations [28]. The Delete in
Breast Cancer-1 (DBC-1) protein is a key regulator of fat storage in
adipocytes mediated by inhibition of SIRT1. In the DBC-/KO mice
exposed to HFD, it was observed high plasma levels of FA with no liver
steatosis, lower expression of senescence cells and increased storage
of FA in the adipocytes with no development of insulin resistance
[28]. Thus, DBC-1 protected liver and adipocytes from senescence
by preservation of the fat compartment function with liver sparing
and insulin sensitivity. Interestingly, a comparison has been made
between this rodent model and the so called ‘healthy obese subjects”
where there is fat accumulation but no signs of metabolic syndrome
or systemic inflammation.
Metabolism and inflammation
Obesity depresses the anti-inflammatory effects of the Heat Shock
Proteins (HSP70) pathway, an inhibition that may contribute to the
progression from NAFLD to NASH [29]. Excess of lipids and fuels
trigger a low grade inflammatory response in both fat and liver tissues
that correlates with the impaired insulin responsiveness. TBARS, a
simple but fair estimate of lipo-peroxidation/malondialdehyde
(systemic oxidative stress) produced throughout the body was shown
to be elevated in plasma from NASH patients when compared to
normal subjects and its levels were correlated with insulin-resistance
status [29]. The former response involved activation of the c-Jun
NH2-terminal Kinases (JNK’s), Endoplasmic Reticulum (ER) stress,
Unfolded Proteins Response (UPR), and the ceramide pathway by
blocking nuclear factor κB (NF-κB) expression at different levels
[29]. In liver tissue, HSP70 down-regulates TNF-α and inducible
Nitric Oxide Synthase (NOS2), genes that increase the inflammatory
response in rodents. In addition, HSP70 in humans induces apoptosis
and increases the concentration of cyclopentenone prostaglandins,
a potent local inhibitor of inflammation. In human liver and fat
tissues, the suppression of HSP70 was strongly correlated with the
upregulation of JNK1 and JNK2 [29]. The authors hypothesized
that the senescence-like state in fat cells have evolved in obese
individuals as an adaptation to the metabolic overutilization of fat
cells, supporting the observation that hepatocyte senescence predicts
NAFLD progression to NASH and to cirrhosis. Patients with different
grades of ESLD from NASH or HCV had significantly decreased
levels of glutathione reduced and increased levels of glutathione
oxidized in plasma when compared to healthy controls [30].
Therefore, a continuous increase in the cell oxidative stress consumes
the antioxidant protective mechanisms and increases the spillage of
oxidative molecules. Increased oxidative compounds accumulation
may induce a progressive larger number of liver cells into senescence
which in turn will enlarge the ASAP component worsening the
inflammatory environment with further increase of stressors into the
liver milieu by triggering an activation of local and systemic immuneregulators
(Figure 3).
Hyperglycemia promotes chromatin remodeling and increased
polyploidy levels in hepatocytes from non-obese diabetic mice; these
alterations are similar but not identical, to the changes observed in
hepatocytes from old mice [31]. Genes involved in glycemic control
and metabolism are also involved in inflammation such as Ppargc1a
(PGC-1α). It acts on the histone deacetylase SIRT1 as a metabolic
sensor in hepatocytes and increases gene activation involved in
the gluconeogenesis pathway [31]. Furthermore, PGC-1α also
plays a role in lipid metabolism [31]. Through the thyroid receptor
pathway, it induces the expression of Srb1 (Scavenger receptor B
member 1), it enhances the uptake of cholesterol esters from High
Density Lipoproteins (HDL) in the liver and inhibits the expression
of Srebp-1 (sterol regulatory element-binding transcription factor-1)
down-regulating fatty acid synthesis [31]. As SASP builds up due
to an increasing number of cells entering senescence, an increasing
insulin resistance state starts to develop with its manifestation,
hyperglycemia which further favors replicative cell arrest.
The fat compartment has emerged not only as an energy
reservoir but as an endocrine organ capable of modulating metabolic
states where the adiponectin/leptin ratio determines an anti or
pro-inflammatory response. Leptin is a 167-aminoacid hormone
expressed predominantly in adipocytes. Its signaling is an important
determinant of food intake, adiposity and energy expenditure [32]. In
the ob/ob mouse, a homozygous mutation in the gene that encodes
leptin is associated with increased appetite, obesity and an insulinresistant
state. When leptin was provided to the ob/ob animal, there
was a dramatic improvement in glucose homeostasis and energy
metabolism. Although leptin related glucose homeostasis is largely
conserved in rodents and humans, most subjects with diabetes and
insulin-resistant states had lower levels of leptin [32]. Adiponectin is
a protein hormone of 244 amino acids synthesized as a monomer of
28 kDa to 30 kDa and assembled in various molecular weights: Low
Molecular Weight (LMW), Medium Molecular Weight (MMW), and
High Molecular Weight (HMW) oligomers [33,34]. HMW oligomers
are the major relevant forms in terms of physiological activities of
adiponectin while, low amounts of HMW oligomers represent an
independent risk factor for several metabolic pathologies such as
obesity-related diseases. Adiponectin plays a pivotal role in energy
metabolism being an insulin-sensitizing hormone and it is involved
in a wide variety of physiological cellular processes including
inflammation, immunity and vascular physiology. Adiponectin
acts through three major functionally distinct and ubiquitously
expressed receptors, AdipoR1, AdipoR2 and T-cadherin. Adiporeceptors
mediate pleiotropic adiponectin actions through signaling
mechanisms involving AMPK, ERK1/2, AKT and P38. In addition,
Polymerase I and Transcription Release Factor (PTRF) regulates
adipocyte differentiation, perhaps fat cell senescence and thus may
determine fat compartment expandability, condition that under
continuous HFD exposure increase the spill-over of FFA to the liver
in combination to a pro-inflammatory adipokine repertoire [35].
Signaling of energy expenditure and metabolism
Rapamycin has an effect in cell life span with significant changes
in the liver transcriptome, effects that are more pronounced when
animals are exposed to caloric restriction [36,37]. In a rodent model,
rapamycin prevented senescent changes with significant differences
by gender but with some common genetic pathways, mainly in the
preservation of mitochondrial function. Those pathways included
protein ubiquitination, NRF2-mediated oxidative stress response
and glucocorticoid and OGF-1 signaling [36,37]. Cell culture studies,
indicated that treatment with rapamycin decreased mitochondrial
membrane potential, decreased O2 consumption, and increased
ATP production [36,37]. Other effects from transcriptome pathways
include a decrease in proteasome activity in parallel with an increase
in cell autophagy, suggesting protein quality improvement processes
and increased resistance to oxidative cell stress effects associated with
reduced cell aging.
The highly conserved Target of Rapamycin (TOR) signaling
pathway is a central regulator of growth and metabolism in all
eukaryotic cells. The mammalian TOR complex (mTOR) encompasses
two structurally and distinct proteins. While the mTORC1 is
associated with anabolic processes such as protein synthesis, lipid
synthesis, nutrient uptake and inhibition of catabolic processes
including autophagy, mTORC2 is insensitive to rapamycin regulated
protein. mTOR2 becomes activated by a family of kinases like the
Serum/Glucocorticoid Kinase (SGK) and Protein Kinase C (PKC).
mTORC1 is up regulated by growth factors, cellular energy status and
is inhibited by the macrolide rapamycin [36-38]. Protein synthesis
is one of the most energy demanding cell functions; a favorable
redox status activates mTORC1 which in turn exercises its actions
down-stream through the ribosomal protein 6 kinase (S6K) and the
eukaryotic translation initiation factor 4E Binding Protein (4R-BP)
[36-38]. Due to the high demand of ATP, mitochondrial function
regulation is of paramount importance on mTOR signaling pathways.
Moreover, mitochondrial dysregulation and continuous mTOR
activation may play a metabolic central role in the transformation and
survival of cancer cells. Mutated cells with malignant potential may
shift their bioenergetic state from ATP mitochondrial production
to cytosol ATP production through the Tricarboxylic Acid (TCA)
cycle. A connection between mTORC2 and mitochondrial function
and cancer appears to be dependent through the HK2 pathway [38].
Nevertheless, recently mTORC2 has been linked to cytoskeleton
regulation through the actin remodeling pathway, which has been
suggested to have an effect on insulin sensitivity/resistance balance
[36,37]. In the tissue specific raptor knockout mice, mTORC1 signaling
in adipose tissue negatively affected whole body energy expenditure,
systemic sensitivity to insulin and thermogenesis [38]. In the same
animal model, mTORC2 signaling in the adipose tissue was a crucial
regulator of liver and pancreas metabolism affecting animal growth
and insulin homeostasis. In addition, mTORC1 and 2 signaling in
the liver affects systemic glucose and insulin homeostasis mainly due
to their effects on Akt and hepatic glucose uptake. Interestingly, liver
tumors in the tissue specific raptor knockout mice, showed a shift
from glucose to glutamine as the main fuel source, making tumor cells
glutamine addictive with high expression of mTORC1 and FGF-21.
Rapamycin treatment may be beneficial as it may inhibit growth on
glutamine addictive tumors. Some liver transplant programs switch
their immunosuppression protocol from tacrolimus to rapamycin
in patients with high risk for HCC recurrence after transplantation.
However and on retrospective studies, its effect on long term overall
survival on patients after liver transplantation for HCC, have had
conflicting results [39,40].
The functional relationship between poly-unsaturated lipid
metabolism, inflammation and cancer development has been discussed
in multiple avenues. Cyclooxygenases (COX’s) and Lipoxygenases
(LOX’s) are enzymatic families that metabolize poly-unsaturated
fatty acids. COX is present in two isoforms (COX-1 and COX-2)
that produce Prostaglandins (PG’s) and thromboxanes, respectively
[41]. LOXs constitute a family of dioxygenases that insert O2 into
poly-unsaturated fatty acids with regional specificity [41]. These
metabolites are biologically active hydroperoxy-eicosatetraenoic
acids that upon reduction form Hydro-Eicosatetraenoic acids
(HETE’s), while the metabolism of linoleic acids preferentially results
in Hydroxyl-Octadecadienoic acids (HODE’s), metabolites known to
modulate inflammation and carcinogenesis [41]. An excess of polyunsaturated
fatty acids could enhance a higher production of HETE’s
and/or HODE’s with an override of pathways that enhances cancer
development. Hepatic COX-2 overexpression induces spontaneous
HCC formation in vitro and in mice through Akt, SKT33 and
mTOR signaling cascades [42]. In the healthy liver, the inhibitor
of the prostaglandin degrading enzyme 15-PGDH potentiates liver
regeneration after partial hepatectomy when compared to control
and sham animals [43]. Thus, prostaglandin active derivatives have
the potential not only to modulate local inflammatory responses
but to promote cell regeneration in the healthy cell and potentially
reversal of cell arrest in the senescent cell.
Metabolism in the liver as a graft
Evidence seems to indicate a peculiar aging pattern for liver grafts
after transplantation. Biological age of the graft does not correspond
to its behavior when transplanted to a different environment of a
younger recipient [44]. One of the most important intracellular
protease systems is represented by the proteasome, the central
catalytic unit of the Ubiquitin-Proteasome System (UPS). No
difference in the accumulation of oxidized proteins and polyubiquitin
conjugates with maintenance of their proteolytic activity was found
in liver grafts after transplantation from younger donors to older
recipient when compared to liver grafts from older donors placed
into younger recipients. Furthermore, there was an increase of the
ß5i/α4 ratio, suggesting a shift toward proteasomes containing
immune-subunits [44]. Thus, it appears older liver grafts transplanted
in younger recipients switched their biological metabolism to
resemble the recipient’s metabolic age. However, the pattern of liver
cell senescence may differ. Liver biopsies, as judged by the senescent
markers telomerase and SMP-30 from older transplanted livers
showed histological damage in asymptomatic patients with up to 43%
and 64% at 5 and 10 years, respectively [45].
In the warm ischemic/reperfusion liver model, Glycogen
Synthase Kinase 3 (GSK-3) ameliorated liver injury upon reperfusion
through an energy-dependent mitochondrial mechanism [46]. GSK-
3 is a serine/threonine kinase regulated by inactivation through
serine phosphorylation. GSK-3 inhibition down regulates the
opening of Mitochondrial Permeability Transition Pore (MPTP)
site, preventing leakage of mitochondrial respiratory chain proteins;
a key step in the activation of caspase dependent apoptosis and
therefore mitochondrial-dependent cell termination. This effect was
present in young animals but abrogated in old animals, and a partial
response was re-established in the older group by glucose infusion
with hepatic glycogen build up storage [46]. Authors speculated that
during reperfusion glycogen degradation provides mitochondrial
fuel in forms of glutamate and α-ketoglutarate maintaining enough
energy levels that preserve mitochondrial membrane integrity or
mitohormesis lowering ROS production, factors needed to decrease
MPTP susceptibility. Former approach in the human was entertained,
where liver graft glycogen replenishment was performed during the
donor phase and evaluated upon reperfusion [47,48]. Metabolic
benefit with improved organ graft function was observed only in
borderline grafts and the ones with high fat content. Nevertheless,
the concept of metabolic replenishment with further graft function
improvement may be refined by strategies of ex-vivo euthermic graft
perfusion prior implantation [49-56].
The chronically diseased liver
Hepatocyte senescence has been demonstrated in chronic
liver disease and as many as 80% of hepatocytes show a senescent
phenotype in advanced liver disease [57]. The effects of insulin
in the liver cell are mediated through two main cellular pathways:
the Phosphatidylinositol 3-Kinase (PI3K)-Akt and the Ras-MAP
Kinase (MAPK) pathways. While both pathways are active in the
regulation of cellular growth, proliferation and differentiation the
PI3K-Akt mediates the metabolic actions of insulin. Those actions
include activation of mTOR1 and its S6 kinase and the inactivation
of Glycogen Synthase Kinase-3 (GSK3) as well as its AS160 with
nuclear exclusion of the Forkhead box protein (Fox01) [57]. In
culture, HepG2 cell lines showed a signaling defect downstream of
the Akt pathway with an impact upon insulin mediated Fox01 cytosol
sequestration and AS160 phosphorylation; a cascade that translated
into insulin resistance of older cells when compared to younger cells.
Maintenance of the senescent state requires Fox01 transcriptional
activity of cell cycle inhibitory genes, even in the presence of growth
factors. Thus, it appears gluconeogenesis and insulin resistance are
unwanted but unavoidable effects of Fox01 gene, which is involved
in cell cycle arrest, detoxification of oxygen species, DNA repair and
gluconeogenesis [57].
FA overload can damage the respiratory chain in the
mitochondrion through a dual role: As an uncoupler and as an
inhibitor [13]. Impairment of the key respiratory state 4→3 can
occur via inhibition of ATP-synthase thereby producing an increase
production of ROS irrespective of ADP concentration. The concept
of Redox-Optimized ROS Balance (R-ORB) postulates that ROS
efflux from the mitochondrion will attain a minimum at intermediate
values of oxidation, when VO2 reaches a maximum following ADP
stimulation. Under state 3 respirations, GSH and thioredoxin systems
are essential for minimizing ROS release from the mitochondria [13].
Moreover, mitochondria from cells with chronic liver disease under
oxidant challenge displayed a two-fold increase in H2O2 emission
when compared to controls along with a 50% decrease in GSH [13].
Since 90% of GSH in plasma is excreted by the liver, glutathione
sp. could serve as a surrogate of cell/mitochondrial stress and their
ratio in plasma may reflect overall liver redox balance [24]. In animal
models of liver malignancy, with or without cirrhosis glutathione
sp (glutathione reduced-GSH, glutathione oxidized-GSSG and
ophthalmate) predicted the growth of malignant cells on normal
livers as early as 14 days after malignant cells implantation and
differentiated animals with cirrhosis by tumor status (HCC+ vs. HCC-
) [58,59]. Furthermore, glutathione sp. in plasma were part of the
metabolic signature that discriminated healthy controls and subjects
after liver transplantation with normal graft function from subjects
with chronic liver disease (Figure 3). In addition, metabolic prints
graded patient’s degree of end stage liver disease which correlated
with the MELD score, and they were able to separate patients with
cirrhosis by tumor status, i.e. HCC+ vs. HCC- [30].
Others argued mitochondrial dysfunction by FA’s respiratory
chain uncoupling is incompatible with thermo-regulatory principles
that governs mitochondrial respiratory chain through energy
demand: intracellular lipids will accumulate whenever FA’s supply
exceeds the energy needs of the cell [13]. While TAG-LD in cells
from a trained individual increases as the source of energy, in the
diabetic obese subject TAG-LD are the result of accumulation with
the subsequent potential overproduction of lipid derived toxins in
the form of LCFA-CoA, Diacyl-Glicerides (DAG) and ceramide,
metabolites responsible, at least in part for the development of
insulin cell resistance [13]. The former theory is attractive in the
heart and skeletal muscle. In contractile cells, optimal excitationcontraction
coupling requires an optimal energy and O2 supply which
in turn affects the Ca2+ handling at the Sarcoplasm Reticulum (SR)
release channels (ryanodine receptors), the SR Ca2+ pumps and the
sarcolemmal Na+/Ca++ exchanger. The heart at rest beats in average
100,000 times per day catalyzing about 6 Kg of ATP to ADP. The
mitochondrion provides the ATP needed for contraction (≈66%)
and the ATP needed for ion transporting (≈33%) essential for the
cardiac electrical activity. Thus, the link among lipid supply and
mitochondrial function, insulin sensitivity/resistance and ion pump
exchange is established for optimal cardiac function or dysfunction
in the obese individual [13]. Perhaps, there is no disputing argument
that lipid oxidation confers a metabolic advantage during starvation
and exercise, but the role of fuel selection per se in defending against
metabolic disease and its role in the liver needs further studies.
In liver, cellular senescence is associated with a pro-fibrogenesis
environment and the relation between advanced liver fibrosis and
shortening of the cell telomere appears to be consistent [11,60].
Telomeres are repetitive DNA sequences (TTAGGG) associated with
the specialized protein shelterin. They are located at the chromosomal
end acting as a cap that stabilizes and protect the chromosome from
erosion and miss-identification as DNA breaks.
During normal cell division, telomeres shorten due to the “end
replicating problem”: The inability of DNA polymerases to fully
replicate the 3’ end of chromosomes [61]. Germline cells overcome
this problem by expressing telomerase, a reverse transcriptase that
maintains telomere length by synthesizing new DNA sequences at
the end of the chromosome [60]. The telomerase complex includes a
reverse transcriptase (TERT) and the RNA component (TERC) [61].
In other somatic cells, continuous cell division results in telomere
shortening which in turn start signaling cell arrest mechanisms,
i.e. senescence or apoptosis. Failure of cell arrest signaling as in
p53 silence sparks further cell proliferation with chromosomal
end-to-end fusions and instability. In addition, exhaustion of liver
regenerative paths and invested mechanisms of telomere repair
could be overcome under continuous and chronic cell injury with
subsequent acceleration of cell senescence and aging. Some studies
had shown that telomere biology is involved in HCC initiation and
its progression [60]. Therefore, telomere shortening is a physiological
marker of cell aging signaling and/or cell arrest preventing further
cell division; failure of cell arrest may end in chromosomal instability
and subsequent mutations may favor tumor development [11,61]. In
fact, the strength of the DNA Damage Response (DDR) in the normal
cell depends ultimately to the degree of p53 gene regulation: A
higher p53 response is associated with apoptosis, a lower response is
associated with cell senescence and a silence p53 response may favor
tumor development and growth [62]. In addition, a sirtuin (SIRT7)
showed an in vivo hyperacetylation of p53 and the SIRT7 knockout
mice suffered among other maladies steatotic liver disease. Sirtuins
were initially identified in yeast as the Silent Information Regulator
(SIR). In mammals, SIRT protein family comprises seven distinct
members involved in cellular survival, senescence and tumorigenesis
[62]. The SIRT7 knockout mice showed a 2.5 fold increase in the liver
triglyceride content and an increased accumulation of hepatocyte
inflammatory markers [62]. Findings that were associated with liver
cells mitochondrial dysfunction through a deacetylate GABPß1
mitochondrial protein pathway and with the development of HCC
through maintaining a deacetylated state of H3K18 at promoters sites
of many tumor suppressor genes [62].
Lipodystrophic syndromes are rare and heterogeneous diseases,
genetic or acquired, where partial atrophy is associated with a
phenotype consistent with insulin-resistant diabetes, dyslipidemia
and NAFLD. Although the genetic causes of these syndromes are
largely unknown, most of the monogenic diseases have in common
primary alterations in the fat tissue consistent with disturbances of the
adipogenesis process or defects in the formation, maintenance and/
or regulation of the lipid droplet [63]. Acquired syndromes are seen
mainly after HIV therapy with anti-retroviral agents as zidovudine
and Stavudine (tNRTl’s). Agents known to render mitochondrial
toxicity with metabolic disturbances similar to the metabolic
syndrome seen in obesity, these metabolic adverse effects include
premature aging associated with impaired prelamin-A maturation
[63]. Lamin-A alterations could produce fragile nuclear envelopes,
alter chromatin organization, increase oxidative stress and promote
premature senescence at the cellular level. The metabolic disturbances
observed in genetic or acquired lipodystrophic syndromes support the
hypothesis of a primary fat compartment dysfunction as the source of
metabolic disturbances, similar to the ones detected in obesity.
Chronic liver disease is associated with an increased translocation
of intestinal bacteria contributory to the liver inflammatory response
and may promote the development of HCC [12]. Liposaccharide
(LPS) produced by Gram + bacteria hosted in intestines from obese
humans and rodents was associated with the transition of NAFLD to
NASH and consequently to its progression to cirrhosis and HCC. LPS
is recognized by the Toll like Receptor 4 (TLR4) which is expressed
upon cell activation on migrating and local macrophages (Kupffer
cells). TLR4 is central for the secretion of TNF-ß and IL-6, cytokines
present in the chronic inflammatory environment that precedes the
detection of malignancy [12]. Further support to the role of LPS
was found by maneuvers such as gut sterilization, removal of LPS
or inactivation of TLR4; maneuvers that diminished tumor growth
in chronically injured livers [12]. In experimental models, dietary or
genetic obesity alterations on the gut microbiota increased levels of
metabolites like Deoxycholic Acid (DCA) that in turn damages DNA.
The enterohepatic circulation may enhance the concentration of such
metabolites further by both encouraging the senescent-associated
secretory phenotype response and favoring a tumor-promoting
environment.
Cellular senescence
Hepatocytes: In response to parenchymal cell loss, hepatocytes
restore the physiological liver mass by self-replication [64]. However,
massive or unending injury may overcome regenerative processes or
may promote a dysfunctional repair process leading to progressive
liver fibrosis, development of portal hypertension and eventually liver
failure. Senescent status was induced in HepG2 cells by exposure to
H2O2. Its consequences and metabolic activity were interrogated [18]
and morphological changes were noted with respect to SA-ß-GAL
and SAF’s expression, cell cycle arrest as well as the upregulation of
p53, p21 and p16 genes. Regarding cytokine expression, IL-8 was
upregulated while IL-6 was downregulated. Disturbances in glucose
and lipid metabolism were evident with upregulation of growth
hormone/IGF1 (SOCS2) and glycolysis (PGM2LT). Nonetheless,
the downregulation of glucogenolysis and gluconeogenesis (G6PC)
was more prominent. The unsaturation of fatty acids was hyperactive
(FADS3) with parallel hypo activity of lipoprotein and hepatic
lipase activity through the Apo-lipoprotein (APC3) system. APC3
also limits the uptake of chylomicrons by the liver cell. Other fatty
acid downregulated proteins included SORL1 (involved in the
uptake of LDL), ACSM2B (a medium-chain fatty-acid-CoA ligase)
and PHGDH indirectly involved in amino-acid synthesis [18]. In
addition, senescent cells secreted a variety of bioactive molecules
including pro-inflammatory cytokines and chemokines that may
influence extracellular matrix and the microenvironment but as well
modulate the immune response with the promotion of macrophage
migration leading to further increase in the inflammatory mielue
[65]. Monocyte Chemotactic Protein (MCP-1) could provide a signal
for monocyte recruitment into the liver followed by activation of
Kupffer cells with the upregulation of death ligands. The expression
of Fas ligand, TNF-α and TNF-Related Apoptosis Inducing Ligand
(TRAIL) further aggravates lipo-apoptosis [66]. In addition the FFA
palmitate increases the expression of TRAIL and abrogation of the
TRAIL receptor expression suppresses the inflammation induced by
nutrient excess in mice [66].
Prior assumptions on cellular senescence determined that cell
cycle arrest was a mechanism to protect the cell towards tumorigenesis.
Nevertheless, it has been shown that the cell in cell cycle arrest can
produce pro-inflammatory mediators, the senescence-associated
secretory phenotype that promotes tumor growth [67]. During
chronic liver disease, senescent machinery becomes “hijacked”
perhaps triggering proliferation and transformation of hepatocytes,
thus, promoting metabolic adaptation which may enhance tumor
grafting and growth [68,69]. The above metabolic paths could at least
in part, be mediated by the over expression of the Phosphatase and
Tensin homologue (PTEN) described in T-leukemia but later shown
in liver tumors to inhibit the Pentose Phosphatase Pathway (PPP)
by binding to Glucose-6-Phophodiesterase (G6PD). With no active
G6DP dimer, cells favor glycolysis with the production of lactate even
in the presence of oxygen [70].
Aging and senescent liver cells have different genetic paths that
may converge to similar metabolic traits. Aging liver cells have a
proliferative response after injury associated with the repression of C/
EBPα, Farnesoid X Receptor (FXR), Telomere Reverse Transcriptase
(TERT), and a decrease in the Wnt signaling pathway [71,72]. A
physiological Wnt signaling pathway involves a soluble ligand that
binds to the Frizzled receptor (Fzd) and the LRP5/6 co-receptor on
the plasma membrane; this interaction activates the cytoplasmic
dishevelled protein which inhibits the ß-catenin (Ctnnb1)
destruction complex (APC, GSK3ß and Axin) by preventing Ctnnb1
phosphorylation and its subsequent destruction. Stable ß-catenin
(intact Wnt signaling) trans locates to the nucleus to form a complex
with Lef and Tcf transcription factors that target genes as c-Myc
and Cyclin D1. In cell culture and a mice model of HCC, tumor
growth was ablated by the suppression of N-Myc Downregulated
Gene1 (NDRG1) expression; it promoted HCC cells to go into cell
arrest [73]. The induction of senescence on malignant cells was
accomplished by upregulation of the tumor suppressor genes p53,
p21 and p16 in addition to decreased phosphorylated Rb. Senescent
liver cells response to injury included transcription of Nf-kB, Myb,
Nkx2-1, Nr5a2 and Ep300 factors; proteins known to be involved in
inflammation, cell differentiation, lipid metabolism and chromatin
remodeling. In addition, the chronic inflammatory phenotype of
senescent cells induces telomere dysfunction and accelerates liver cell
aging [74]. Thus, decreased physiological cell signaling that occurs
with aging plus stress induced cell senescence may add to the lipid
toxic microenvironment by promoting a vicious circle that overrules
redundant mechanisms that prevent uncontrolled cell division.
Mechanisms that imply both an apoptosis ‘switch” from a proapoptotic
to an anti-apoptotic status and a role of the Bcl-2 proteins
family to determine cellular fate [75].
Cellular event that follows are the activation/repression of
factors involved in cell proliferation. In the liver cell, the known
transcriptional shift includes activation of FOXO3, FOXII, E2F1,
c-jun, C/EBPß, Myb, USF and neutralization of inhibitors of cell
proliferation such as Rb family and C/EBP family of proteins
[76]. In C/EBP-S193A mice, failure to stop liver regeneration after
surgery correlated with the epigenetic repression of C/EBPß, p53,
FXR, SIRT1, PGC1α and TERT. The repression was performed by a
protein formed by C/EBPß-HDCAC1 complex which also inhibit the
promoters of enzymes for glucose synthesis PEPCK and G6P [76].
The response of cell cycle engaged hepatocytes and cell cycle arrested
hepatocytes (senescent cell) to injury is different and it may awake an
unregulated cell growth on quiescent stem liver cells [76,77].
Cholangiocytes: Cholangiocytes are elongated cells that cover the
path of actively secreted hepatocyte’s bile, a fluid that is conducted,
and modified in bile duct radicals that finally reaches the extrahepatic
biliary ducts. Although cholangiocytes are metabolically very active
cells involved in the secretion and resorption of water and soluble
bile components, they are not directly involved in the metabolism
and/or regulation of biliary lipid species (cholesterol, bile acids and
phosphatidil-choline vesicles) [78,79]. Oval shaped liver cells may
differentiate into cholangiocytes with a distinct metabolism and
perhaps pathway towards malignancy [15,16]. However, obesity
and hepatitis B/C virus infections are not associated with a higher
incidence of cholangiocarcinoma.
Hepatic stellate cells (HSC) and Portal myofibroblasts
(MF)
HSC are quiescent cells that express typical markers of both
neural cells and adipocytes (glial fibrillary acid protein-GFAP,
peroxisome proliferator-activated receptor gamma-PPA, and
adiponectin receptors). They are activated by cytokines, growth
factors, ROS, damaged cells and apoptotic bodies [64]. In health, MF
is located adjacent to bile duct epithelia and is the first responder
to biliary injuries. Upon activation HSC’s acquire a MF phenotype,
cells that upon phagocytosis of LD and/or apoptotic bodies from
damaged cells get additional energy and became Fas-ligand and
TNF-α unresponsive to apoptosis; mechanism in use for increase
collagen synthesis and deposition [64]. Furthermore, activation of
the adenosine receptor A2A increases HSC proliferation and inhibits
death and senescence by down regulation of p53 and Rb through the
cAMP-PKA/Rac1/p38 MAPK pathway [80]. Activated MF’s express
CCN1/CYR61, an important regulator of inflammation and wound
healing. Cystein-rich 61-protein (CCN1/CYR61) is a matrix-cellular
protein that induces senescence at later stages of wound healing
by promoting tissue remodeling through fibrogenic cell apoptosis
and attenuation of TGF-ß signaling [81]. HSC and MF senescent
fibrogenic cells no longer proliferate, thereby reducing the load of
ECM deposition. In addition, senescent fibrogenic cells express an
increase in the secretion of Metalloproteinases (MTP’s) leading to
matrix degradation. Apoptotic fragments from HSC and MF are
cleared by natural killer cells promoting wound healing, the best
characterized mechanism of fibrogenesis resolution [64,81]. NFκB
is a key regulator for HSC survival and proliferation by maintaining
the expression of Mcl-2. Inhibition of NFκB increases HSC apoptosis
by up-regulation of the JNK pathway. Thus, the activation as well as
the induction of senescence/apoptosis of HSC/MF is normal wound
healing mechanisms that promote the establishment of normal organ
architecture and function with clear paths of initiation and resolution.
During chronic cell injury, such as in a state of high caloric intake
enriched with lipids, an increase and progressive pool of biologically
active HSC’s may become prominent [11]. An incremental
chronic state of fibrogenesis alters hepatic architecture leading to a
concomitant increase in portal flow resistance, portal hypertension
and the development of collateral circulation. In addition, HSC’s
produce a microenvironment with altered Extracellular Matrix
(ECM) that provides biochemical and mechanical cues to the growth
and establishment of tumor cells [67]. Nevertheless, since 90% of the
HCC’s flourish in a highly progressive fibrotic ECM, the question
raises if it is the changes on the microenvironment that further
promotes metabolic transformation with an “apoptotic switch” and
tumor development. Interestingly, progressive liver fibrogenic ECM
becomes enriched with Vascular Growth Factor (VGF) receptor
promoting angiogenesis, paving the way for the much needed arterial
high O2 supply for HCC expansion [67].
The different components of the ECM, cellular and non-cellular
interact directly and indirectly with malignant cells therefore
changing the phenotype of the evolving cells that in turn produces
feedback signals to further adapt the microenvironment to the needs
of the malignant cell. The mechanical stress arising from the ECM
constitute the “out-side-in” signaling linking the actin cytoskeleton
to the microenvironment by increasing intracellular contractile
forces regulating signaling pathways fundamental to determine cell
phenotype. In response, the anchored cells expressed adhesions
molecules and secreted proteins that signals HSC and other ECM
regulators increasing anchoring sites in response to the “in-sideout”
signaling [67]. Therefore, the metabolic transformation of the
already stressed parenchymal cells help to choose a path different to
senescence and necrosis but to a path of unregulated regeneration,
thus escaping apoptosis. A path, that needs an ECM differentiation to
assure cell survival in a non-efficient energy redox status.
Sinusoidal endothelial cells (SEC)
SEC’s are specialized endothelial cells that lie flat in the liver
sinusoids along and in direct contact with the hepatocytes. Through
their membranes and specialized pores or fenestra passes high
concentrations of metabolites, proteins and other blood compounds,
traffic which is regulated by the size of the fenestra. SEC’s play a
critical role in immune-activation, rolling of T cells, macrophages
and PMN migration. Liver sinusoidal endothelial cells may be
affected with age and obesity. SEC from old individuals have impaired
and reduced expression of VEGF likely due to impaired nuclear
transport of P-STST3 and P-CREB transcription factors [82,83].
In a rodent model of sepsis, endothelial Nitrogen Oxide Synthase
(eNOS) deficient mice and aging mice had the same mortality and
mitochondrial dysfunction upon the isolation of SEC mitochondrion
[84]. In obesity and during early fibrogenesis, SEC loses their fenestra,
decreasing the exchange of metabolites and increases the secretion of
several basement membrane components (type IV collagen, perlecan,
entactin and laminin) [64]. Authors concluded that an endothelial
base-line dysfunction in the aging animal is manifested by a
weakened antioxidant response and inappropriate energy production
from mitochondrial dysfunction due to a tipped-balance of the SEC
oxi-redox systems when exposed to additional stress. This is seen
in the obese towards a state of energy depletion and cellular death,
apoptosis or activation of a pro-coagulant/pro-fibrogenic phase. The
changes of SEC’s with aging may limit O2 delivery and availability
to liver cells with its potential effects on mitochondrial function, a
pro-fibrogenesis state and the promotion of insulin resistance status.
Changes exaggerated in obesity, implying obesity may promote
accelerated SEC aging processes. Interestingly, endothelial cellular
senescence was inhibited in vitro and in the rodent by the activation of
the Liver X Receptor (LXR), a nuclear receptor involved in the control
of hepatic lipid and cholesterol metabolism [85]. Furthermore, LXR
has been shown to play an important role in glucose metabolism,
cytokine production and anti-inflammatory response.
Three types of SEC’s co-exist in the normal liver sinusoid: Mature
SEC, SEC progenitors and bone marrow-derived SEC progenitors
[86]. Mature SEC is gatekeepers of fibrogenesis by maintaining
HSC in their inactivated state. SEC’s regulate sinusoidal blood flow
through their action on HSC and thus keep a low portal pressure [86].
In addition, mature SEC’s have the largest endocytic capacity in the
body fulfilling their dual cell clearance capacity (from the arterial/
systemic and portal/gut systems). The liver endocytic function has
been implicated in a liver-renal axis where the lack of SEC-stabilin-2
receptors inhibit the clearance of toxic molecules that manifest
with mild liver fibrosis without liver dysfunction but with renal
glomerular fibrosis. Not only do SEC’s have many glycoproteins that
serve as receptors for bacterial epitopes but as receptors for immunemodulation
and pro-coagulant activity. The above mentioned SEC
functions are at least partially lost at the time of sinusoid capillarization
[86]. SEC capillarization is characterized by the disappearance of the
fenestrae, development of a basement membrane and the appearance
of characteristic markers. This phenomenon happens in chronic
liver injury and it precedes activation of HSC and sequestration of
macrophages. The angiogenesis process that follows is mediated by
VEGF, an angiocrine response that drives neo-vessel formation in
direct proportion to the degree of the sinusoidal pressure gradient.
Furthermore, SEC pseudo-capillarization refers to changes that
occur in endothelial cells associated with aging and senescence. It is
manifested by a decrease of up to 50% of their fenestrae, development
of a patchy basement membrane and partial SEC dysfunction [86].
Chronic exposure of high fat diet may accelerate aging/senescence of
SEC, endothelial dysfunction with recruitment of systemic immune
cells and activation of Kupffer cells inducing HSC into a fibrogenic
state followed by an angiocrine response that decreases hepatic blood
flow, O2 delivery, and clearance of toxic molecules. As metabolic
stress of neighbor hepatic cells already in mitochondrial distress due
to fat accumulation progresses, a constant and growing inflammatory
milieu enhances tumor development, immune-recognition failure
and then progress and spreading of malignant cells.
Interestingly, aging endothelial cells from the fat compartment
of mice was associated with adipose dysfunction manifested by
ectopic (liver) fat deposition and adipose tissue fibrosis, increased
adipose mitochondrial oxygen flux, altered lipid utilization, increased
tissue oxidative stress and lower gene expression in visceral fat [87].
Nevertheless, and most important, these findings were associated
with reduce fat tissue vascularity, reduced angiogenic capacity and
endothelial dependent dilation with reduced Nitric Oxide (NO)
bioavailability [87]. Limited oxygen mitochondrial availability
contributes to the pro-oxidative older adipose tissue phenotype that
can further impair both insulin action and vascular function, a key
element in local and systemic insulin-resistant related metabolic
syndromes. Changes that are exaggerated in obesity, implying obesity
may promote accelerated aging processes in many organs.
Resident liver immuno-cells
The anatomical location of the liver and its dual blood supply
ensures an optimal exposure of antigens to the hepatic resident
immune cells not only from nutrients and GI microbiota but from
systemic compartments, such as the adipose compartment. Kupffer
cells in concert with NK, CD4+T-cells, and local antigen presenting
cells modulates the liver immune status. Kupffer cells constitute 80%
of the tissue fixed macrophages and 20% of the total cell population
of a normal liver [9]. Their characteristic macrophage activity is
polarized mainly in portal tracts where the antigen dynamics is higher
from food and bacteria. Innate macrophages have the potential to
initiate an inflammatory response of different proportions by upregulating
adhesion molecules such as ICAM-1, and cytokines as
TNF-α, IL-1, IL-6, MIP1α, TGF-ß and RANTES. Activation that
can only lead to antigen presenting cell to cell communication and
amplification and enrichment of the microenvironment with ROS
promoting subsequent parenchymal cell apoptosis/necrosis. Natural
Killer (NK) and CD8+T-cells developed a specific signature in livers
with NASH from mice under HFD [88]. The depletion of CD8+Tcells
protected murine from NASH progression but not from weight
gain. In addition, NK T-cells in the liver express markers that
recognize lipid antigen CD1d [9]. Liver NK cells undergo Thymus
clonal double deletion but are positive for CD3 and CD56 and
they were thought to be CD1d independent. Nevertheless, hepatic
antigen-presenting cells may introduce microbial glycolipid antigens
to NK cells, stimulating secretion of Th1 or Th2 cytokines which
subsequently initiates an adaptive response. Hepatic NK cells have as
well the ability to secrete osteopontin and sonic hedgehog, molecules
known to promote the transition from NAFLD to NASH [9]. The
most accepted hypothesis, continuous cell parenchymal damage and
necrosis adds to a chronic inflammatory environment a dysregulation
of the cell cycle regenerative process rendering tandem mutations and
thus malignant cells was challenged [89]. On the NEMO knockout
mouse, authors were able to develop HCC through a death receptorindependent
FADD signaling pathway. Nevertheless, it wasn’t until
recently that the link between a metabolic hostile microenvironment,
immune-recognition failure and HCC presence was established [90].
An increase in liver tissue of linoleic acid, a FA selectively toxic to
CD4+ T-cells had been significantly associated with low CD4+ T cells
counts and the presence of HCC, in the rodent and human models.
CD4 (+) T lymphocytes have greater mitochondrial mass than CD8
(+) T lymphocytes and generate higher levels of mitochondrially
derived ROS. Disruption of mitochondrial function by linoleic acid,
a fatty acid accumulated in NAFLD, causes more oxidative damage
than other free fatty acids such as palmitic acid and mediates selective
loss of intrahepatic CD4 (+) T lymphocytes. Local metabolic changes
could alter the immune response to a one that favors malignant cell
expansion.
In the obesogenic environment, aberrant activation of immune
cells has emerged as key features of the metabolic syndrome. The
interaction between the adipose compartment and the liver tissue
has been hypothesized as a critical interface for nutrient sensing and
metabolic control [9]. In the rodent model, neutrophils infiltrate the
adipose compartment as early as three days after starting a high fat
diet, however its role as well as the role of basophils and eosinophil
cells has not yet been clarified. Mast cells, which have been observed
in increasing number, have been implicated in the secretion of IL-6
and IFN-Ϫ [9]. Moreover, leptin, a hormone secreted specifically by
adipocytes has been found to be increased during high fat diets and
upregulated the expression of leptin receptors on NK T-cells. This
regulation is time sensitive, and chronic leptin stimulation change NK
cells from an inflammatory like response to a dampned one, favoring
at long term, in the liver and perhaps in other organs a susceptibility
to low recognition of no self-cells, impaired anti-tumor surveillance
and a nest for the flourishing of cancer. The former hypothesis finds
support in the obese mice, where it was observed a switch from the
normal Th1 immuno-response to the Th17 immuno-repertoire,
phenotype that deteriorates autoimmunity [9].
Extracellular matrix (ECM)
The Extracellular Matrix (ECM) is formed by a non-cellular
component in tissues and organs composed primarily of water,
proteins and proteoglycans. Components created an intricate
scaffold where organ cells get structural support with a dynamic and
continuous traffic of water, ions, metabolites, proteins and cells on
passant to maintain organ physiology. As such, ECM interactions
with organ cell components regulate cell differentiation, adhesion,
proliferation, migration and survival [64]. The collagen family is
the major fibrillar proteins of the ECM and the body (approx. 30%
of the total protein contain) [64]. There are three main classes of
collagen, fibril-forming which include types I, II, III, XI, XXIV
and XXVII the most common varieties and their role is mainly
mechanical by conferring tensile strength to both tissue and organs.
Fibril-Associated Collagens With Interrupted Triple helix (FACIT’s)
includes type IX, XII, XIV, XIX, XX, XXI and XXII; this subclass
of proteins don’t form fibrils themselves but bind to the surface of
pre-existing collagen favoring fibril enlargement. Finally, type III
collagen serves as anchoring collagen between the epithelial cells
and the lamina reticularis constituting the basement membrane
where type IV collagen is most abundant. Non-collagenous proteins
include fibronectin, tenascin, laminins, fibrillins and matricellular
proteins. While the former peptides play a major role in cell
differentiation, cell growth, adhesion and migration, matricellular
proteins, i.e. thrombospondin-1 and 2, osteonectin, osteopontin and
cyr-61/Connective Tissue Growth Factor (CTGF) serve mainly as
a vehicle for cell signaling. Proteglycans are carbohydrate enriched
proteins which retain large quantities of water regulating the smooth
trafficking of molecules to and from the cell with numerous signaling
active sites for growth factors.
The ECM continuous remodeling is a complex process that
integrates proteins and cellular components from local and distal
environments [64]. The degradation of ECM proteins is closely
controlled by Matrix Metalloproteinases (MMP’s), a superfamily of
zinc-dependent endopeptidases highly regulated by specific inhibitors
such as the Tissue Inhibitor of Metalloproteinases (TIMP’s). In the
liver, cellular component involved in collagen synthesis and deposition
included HSC, MF and vascular smooth muscle cells [64]. In chronic
liver injury, an override mechanism of collagen deposition regulation
promotes massive ECM expansion. The characteristic features of
abnormal liver fibrogenesis as a consequence of continuous liver
injury and activation of collagen secreting cells include damage to
the epithelial/endothelial barrier, recruitment of inflammatory cells,
secretion of cytokines and other inflammatory mediators, further
generation of ROS, progressive deposition of collagen with expansion
of ECM and worsening organ fibrosis and subsequent metabolic
changes of portal hypertension.
Mitochondrial senescence
The mitochondria, a double membrane cell organelle varies in
number and its presence is linearly associated with the metabolic
activity of the organ and its required energy requirements in form
of ATP. Within the mitochondrial matrix a series of biochemical
reactions occur. Acetyl-choline primer is reduced through the
tricarboxylic acid cycle converting glycolysis-derived pyruvate into
NADH and succinate. The former compounds couple another set of
reactions at the inner membrane border where the Electron Transport
Chain (ETC) is present to boil an oxidative phosphorylation process.
The ETC is composed of five enzymatic complexes (I to V; NADHCoQ,
succinate-CoQ, CoQ-cytochrome reductases, cytochrome c
oxidase and ATP synthase, respectively) where NADH is the substrate
of ETC-C1 and succinate the substrate of ETC-CII [10]. After
oxidation, electrons are transferred from Complex I to CII to CIII
and finally to Complex IV where oxygen is reduced to form H2O. The
electron transport process is coupled to a proton pumping process
creating a proton gradient between the mitochondrial membranes,
gradient that is dissipated by Complex V (ATP synthase) through
ATP synthesis. A control mechanism is created by the “proton leak”,
mechanism that generates heat instead of ATP [10]. Much of the leak
is a catalytic reaction generated by the Uncoupling Proteins (UCP’s)
which play an important role in reducing proton gradient, heat and
ROS [10]. Mitochondrial aging and senescence are linked to reduced
ATP production and increase ROS production, i.e. Superoxide (O2-
), Hydrogen Peroxide (H2O2) and Hydroxyl radical (OH-) which
are mostly produced because electron leakage at the level of CI and
CIII [17]. Mitochondria function benefit from the role of estrogen
in plasma through it's binding to the nuclear estrogen receptor that
enhances a signaling to prevent oxidant stress and also inhibits the
renin-angiotensin-aldosterone system [10]. Thus, sex differences in
mitochondrial function may explain the disparity in overall survival
between men and women, differences that may be taken into account
during animal models studies.
The reasons why the mitochondrion conserves a cell independent
genome are not clear, but it is intuitive to imply self-energy regulatory
processes are united through a fine tune mechanism between energy
expenditure (ATP use) and energy production (ATP synthesis)
at every organelle level. It may provide an overall advantage for
survival of the cell, the organ and entire biological living system.
The gradual ROS response theory of aging argues a protective role
of ROS in early life, when cell oxidative damage and ROS production
are low; however, later in life ROS reaches a level where its beneficial
effects (as the one observed in dietary restriction and/or exercise) are
overcome by its detrimental effects elicited by a higher cell oxidative
stress (as the one observed in high fat diet and sedentary habits) [18].
The effects that are amplified include loss of genomic controls (p53),
microRNA dysregulation, loss of function of Telomerase Reverse
Transcriptase (TERT) and a lower immune-surveillance status.
Although the role of p53 in the mitochondrion is not completely
clear, p53 binds to the Peroxisome proliferator-activated receptor
Gamma-Coactivator1 alpha and Beta (PGC-1α and ß) fomenting
their inhibition of expression and therefore downregulated oxidative
function. In addition, p53 target p16 and p21, factors that triggers G1-
phase cycle arrest by inhibiting cell cycle regulatory kinases Cdk4 and
Cdk2 [18]. The third known effect of p53 at the mitochondrion level
is to promote cell apoptosis by increasing mitochondrial membrane
permeability with leakage of cytochrome proteins, a direct activator
of the caspase cascade. The function of TERT is highly affected by
levels of ROS production and its protective patterns are only observed
with low ROS levels. The role of microRNA in the mitochondrial
environment remains to be elucidated.
The Mitochondrial Free Radicals Theory of Aging (MFRTA)
has been the most popular theory to explain the cell aging process
where increasing production of mitochondrial ROS with lower
ATP production are the main factors responsible for cell aging and
corresponding mitochondrial ultra structure changes [17,91]. As
mentioned, leakage of electrons at the level of CI and CIII transfer are
larger with age and the higher potential for DNA damage. 8-oxo-7.8-
dihydro-2’deoxyguanosine (8-oxodG) is one of the most abundant
DNA mutations caused by oxidative conversion to guanosine.
Furthermore, its accumulation follows an inverse and exponential
curve against life expectancy in several mammals [17]. Recently,
it was described that humans with longer longevity have a higher
content of mitochondrial DNA (mtDNA) per cell in different organs,
and support the notion of ethnic background on mtDNA influence
and life span. The frequency of mtDNA mutations occurs at different
rate depending on the organ. Skeletal and cardiac muscles, liver and
kidney are more affected by somatic mtDNA mutations compared
to other organs such as the skin and lung [17]. Furthermore, the
clonal expansion of mtDNA mutations occurs via a phenomenon
called genetic drift, a random propagation and expansion of DNA
mutations occurring at each DNA replication. The drift of mutations
may be more important in metabolically more active organs that
require more energy expenditure and therefore more ATP synthesis.
The expansion of mtDNA mutations may be enhanced not only by its
duplication and drift but also by a lower state of DNA damage repair
mechanisms [17]. The Base Excision Repair (BER) process is impaired
in senescence and aging due to a loss of function to BER associated
proteins CSA and CSB. Thus, the increase production of ROS creates
a vicious loop of mtDNA mutations than in turn favor an increase
production of ROS perpetuating and enhanced organelle dysfunction
by defective reparative mechanisms. A naturally occurring thymidine
to cytidine mutation in the mitochondrial stressors tRNAILE gene
is associated with phenotypes of hypertension, hypercholesterolemia
and hypomagnesaemia [10]. Furthermore, the DNA A3243G
mutation causes impaired insulin secretion and polymorphisms in the
promoter of the UCP2 protein, alterations associated with increased
incidence of obesity, reduced insulin secretion and DMII [10].
Mitochondrial function may be impaired in chronic high fat
diet challenge as a result of a decrease in ß-lipid oxidation. Indirect
evidence showed an accumulation of diacylglycerol and fatty-
Acyl-CoA which in turn activates stress-related serine/threonine
kinase activity and inhibits glucose transport [10]. Oxidative stress
contributes further to impaired insulin signaling increasing UCP2
activity which in turn enhances “proton leak” with uncoupling of
the glucose metabolism pathway and decreased ATP production. A
progressive higher lipid peroxidation may favor further oxidative
stress with DNA damage and low DNA repair by affecting members of
the Bcl-2 family, triggering an influx of Ca2+ with subsequent opening
of the mitochondrial permeability transition pore, cytochrome-c
leakage to the cytosol and activation of the caspace-3 complex. Cell
self-digestion and nuclear DNA fragmentation overcomes with the
typical cell fragments morphology [10]. Alternatively, DNA damaged
and telomerase shortening results in mutations that may affect
mitochondrial function to a level of organelle survival but inefficient
ATP production assuring the “apoptosis switch” and diverting
biochemical reactions to a cytosolic site for ATP production. The
later assumption may find some support in the observations that
tumor development and early growth is favored in low O2 delivery
zones and that tumor development is associate with increase lactate
production, the Warburg effect [30,69].
Future directions
Prevention of metabolic syndrome and its health consequences is
primordial. A healthy diet that is balanced not only in calories but also
in its components, specially fats and carbohydrates would avoid fat
storage spillage from a saturated fat body compartment. In addition,
a substantial use of lean mass through directed exercise will decrease
further cell, organ and body aging. The central nervous melanocortin
system forms a neural nutrient sensing network connecting signals
of metabolic state with centers of the brain that regulates intake
behaviors and metabolic homeostasis. Central administration of
α-MSH reduces food intake and may also increase energy expenditure
resulting in weight loss [92]. Metabolic disturbances in the liver
render liver cell changes that progress from NAFLD to NASH to
cirrhosis and malignancy. A non-invasive plasma based monitoring
of such changes on disease progression and treatment response as
well as for tumor screening may be possible by metabolomic liver
prints in the near future [30].
There have been a myriad or reports on compounds that not only
prevent but reverse cell aging and some even malignant development
in the animal model [25,93-102]. Curcumin, the major bioactive
compound of turmeric spice, through its antioxidant and antiinflammatory
properties has been claimed to retard tumorigenesis
and diabetes and to modulate lipid metabolism [103]. Furthermore,
curcumin prevents the development of atherosclerosis and NASH,
perhaps by the upregulation of a fatty acid binding protein present
in adipocytes (aP2) but also found in macrophages (FABP-4). This
protein is a cytosolic protein present in adipocytes and macrophages
which modulates the trafficking of lipids/cholesterol processes and
activation of inflammatory mechanisms through CD36 upregulation
and reduced expression of NF-kß thus, decreasing cytokine secretion
[103]. Prior studies showed that high fat diet and obesity promoted
liver tumorigenesis by inducing chronic inflammation through
the IL6/STAT3 pathway and, STAT3 activated tumors has been
showed to be more aggressive in humans. Lycopene attenuated HCC
occurrences in the animal model through downregulation of the
STAT3 signaling [95]. The aqueous extract of Ligustrum lucidum
fruit induced apoptosis through the activation of the caspase cascade
and cellular senescence by upregulation of p21 and downregulation
of RB phosphorylation [102].
Other molecules with promising cell aging and tumor repression
properties included the COX-2 and a Na/K/ATP signaling
mechanisms. Inhibition of 15-Hydroxyprostaglandin Dehydrogenase
(15-PGDH), a prostaglandin-degrading enzyme, potentiates tissue
regeneration in multiple organs in mice [43]. In a chemical screen,
authors identified a small-molecule inhibitor of 15-PGDH that
increases prostaglandin PGE2 levels in bone marrow accelerating
hematopoietic recovery in mice receiving a bone marrow transplant.
It also promoted tissue regeneration in mouse models of colon and
liver injury. Selective COX-2 products may have rescued telomere
dysfunction, cell senescence and tissue regenerative potential [74].
However, its mechanism and signal transduction remains to be
determined. pNaKtide is a synthetic peptide that conserves the
active sequence for the ligand-binding capacity to the ß-subunit of
the transmembrane Na/K-ATPase. Although the Na/K-ATPase
mainly exercise its function as an ion exchanger pump vital for
cell survival, recently it was shown to elicit nuclear signaling that
regulates mitochondrial function and cell energy production through
a Src/ERK pathway [104-112]. Furthermore, pNaKtide prevents the
development of atherosclerosis and fatty liver disease in the HFD
mice model with significant amelioration of ROS. In addition, it
down-regulates collagen synthesis and inhibits growth of human
cancer cells in vitro. Translation of promising compounds to the
treatment of patients with NAFLD/NASH is expected in the near
future to further prevent the consequences of advanced liver fibrosis
and HCC development.
Figure 1
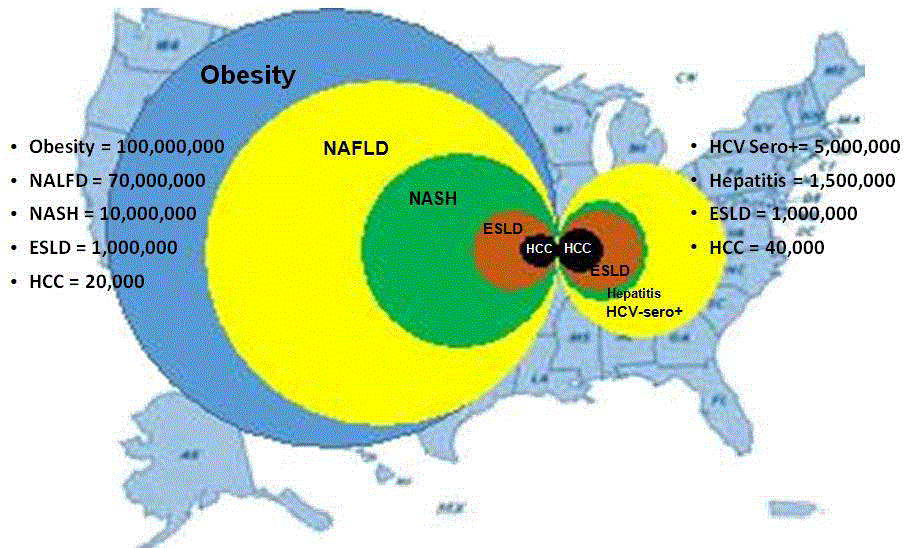
Figure 1
Main causes of End Stage Liver Disease (ESLD) and Hepatocellular Carcinoma (HCC) in USA. Hepatitis C Virus infection (HCV) and obesity are
the main cause of the Global and Western increase in ESLD and HCC. While prevention relays in stopping virus transmission and implementing programs
of healthy caloric intake and exercise programs, treatments of established ESLD and its malignant consequence are similar. Nevertheless, the most effective
surgical treatment, liver transplantation is limited due to scarcity of donors and loco-regional therapies have limited survival effect due to malignant recurrence or
progression of liver dysfunction.
Figure 2A

Figure 2A
Morphological changes observed in the mice model of High Fat Diet (HFD) plus fructose (Western Diet) in the microbiota, fat content tissue and liver.
Liver cells accumulate FA in form of TG from the spillover of lipid excess in the fat compartment and after saturated the normal processes of liver lipid metabolism.
Figure 2B

Figure 2B
Local liver inflammatory response from lipid excess and cell involved in the hepatic ecosystem. Lipotoxicity an addition to increase LPS activates SEC,
HSC and Kupffer cells inducing more parenchymal cells into senescence and apoptosis and change of the local mielue into an inflammatory microenvironment.
Continuous HFD decreased further mitochondrial function with lower ATP production, increase collagen deposition and progressive liver fibrosis, liver dysfunction.
The state of progressive liver fibrosis due to a local and systemic inflammatory state results in an increasing insulin resistance status with the full metabolic
syndrome phenotype. Its progression results in decompensated ESLD and the development of malignancy.
Figure 3
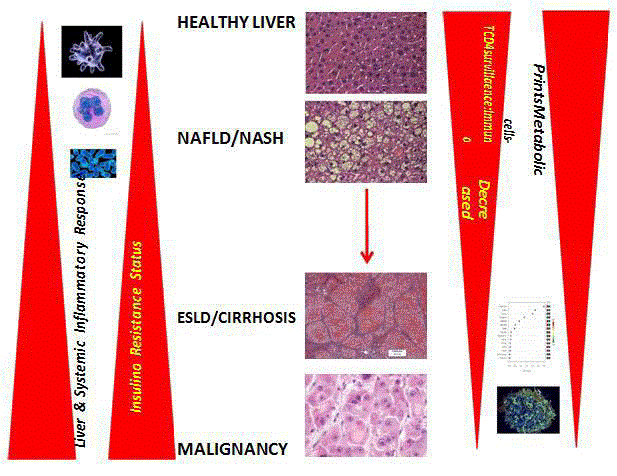
Figure 3
Local and systemic responses that occur in the progression of NAFLD to NASH and ESLD associated HCC and their metabolomic print. The Liver local
progression from NAFLD to NASH is associated with a local inflammatory response that eventually involves several other organs and becomes systemic. The
systemic inflammatory response is associated with the development of an insulin resistance status and the metabolic syndrome phenotype, HTN, central obesity
and DM Type II. Continuous liver lipotoxicity decreased mitochondria function, decrease ATP production and enhance the secretion of lipid intermediates toxic
to CD4 T lymphocytes. All together enhances a process of regenerative stimulus of in majority senescence cells with mitochondrial dysfunction generating at
some point a metabolic swap of ATP production to the cytosol, which may be associated with a mitochondrial generated apoptotic switch and in an environment
of progressive fibrosis and therefore low oxygen and nutrients delivery favoring the survival of the already highly mutated cells which in turn have escaped
physiological cell cycle control and immuno-recognition assuring cell clone growth. Metabolic disturbances precede cell cycle variation and genetic expression
creating metabolic signatures of liver status in health and disease.
References
- Collaborators GBDRF, Forouzanfar MH, Alexander L, Anderson HR, Bachman VF, Biryukov S, et al. Global, regional, and national comparative risk assessment of 79 behavioral, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386(10010):2287-323.
- DALYs GBD, Collaborators H, Murray CJ, Barber RM, Foreman KJ, Abbasoglu Ozgoren A, et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145-91.
- Global Burden of Disease Cancer C, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;1;3(4):524-48.
- Global Burden of Disease Cancer C, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-LM, et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1(4):505-27.
- Global Burden of Disease Pediatrics C, Kyu HH, Pinho C, Wagner JA, Brown JC, Bertozzi-VA, et al. Global and National Burden of Diseases and Injuries Among Children and Adolescents Between 1990 and 2013: Findings From the Global Burden of Disease 2013 Study. JAMA Pediatr. 2016;170(3):267-87.
- Khachatryan V, Sirunyan AM, Tumasyan A, Adam W, Bergauer T, Dragicevic M, et al. Search for dark matter, extra dimensions, and unparticles in monojet events in proton-proton collisions at [Formula: see text][Formula: see text]. Eur Phys J C Part Fields. 2015;75(5):235.
- Stanaway JD, Flaxman AD, Naghavi M, Fitzmaurice C, Vos T, Abubakar I, et al. The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013. Lancet. 2016;388(10049):1081-8.
- Collaborators GOaO. Obesity and overweight and their health impact 1990-2015 in 195 countries. N Engl J Med. 2017.
- Eheim A, Medrikova D, Herzig S. Immune cells and metabolic dysfunction. Semin Immuno pathol. 2014;36(1):13-25.
- Jia G, Aroor AR, Sowers JR. Estrogen and mitochondria function in cardiorenal metabolic syndrome. Prog Mol Biol Transl Sci. 2014;127:229-49.
- Dongiovanni P, Romeo S, Valenti L. Hepatocellular carcinoma in nonalcoholic fatty liver: role of environmental and genetic factors. World J Gastroenterol. 2014;20(36):12945-55.
- Karagozian R, Derdak Z, Baffy G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism. 2014;63(5):607-17.
- Aon MA, Bhatt N, Cortassa SC. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol. 2014;5:282.
- Huppert SS, Campbell KM. Emerging advancements in liver regeneration and organogenesis as tools for liver replacement. Curr Opin Organ Transplant. 2016;21(6):581-7.
- Alison MR, Lin WR. Regenerating the liver: not so simple after all? F1000Res. 2016;5:F1000.
- Lisman T, Porte RJ. Mechanisms of platelet-mediated liver regeneration. Blood. 2016;128(5):625-9.
- Lauri A, Pompilio G, Capogrossi MC. The mitochondrial genome in aging and senescence. Ageing Res Rev. 2014;18:1-15.
- Aravinthan A, Shannon N, Heaney J, Hoare M, Marshall A, Alexander GJ. The senescent hepatocyte gene signature in chronic liver disease. Exp Gerontol. 2014;60:37-45.
- Carr SK, Chen JH, Cooper WN, Constancia M, Yeo GS, Ozanne SE. Maternal diet amplifies the hepatic aging trajectory of Cidea in male mice and leads to the development of fatty liver. FASEB J. 2014;28(5):2191-201.
- Chen J, King K, Zhang JX. Effect of caloric restriction on hepatic sinusoidal system and stellate cells in mice. J Aging Res. 2014;2014:670890.
- Bozhkov AI, Nikitchenko YV. Thermogenesis and longevity in mammals. Thyroxin model of accelerated aging. Exp Gerontol. 2014;60:173-82.
- Uebi T, Umeda M, Imai T. Estrogen induces estrogen receptor alpha expression and hepatocyte proliferation in the livers of male mice. Genes Cells. 2015;20(3):217-23.
- Cechella JL, Leite MR, Dobrachinski F, da Rocha JT, Carvalho NR, Duarte MM, et al. Moderate swimming exercise and caffeine supplementation reduce the levels of inflammatory cytokines without causing oxidative stress in tissues of middle-aged rats. Amino Acids. 2014;46(5):1187-95.
- Kombu RS, Zhang GF, Abbas R, Mieyal JJ, Anderson VE, Kelleher JK, et al. Dynamics of glutathione and ophthalmate traced with 2H-enriched body water in rats and humans. Am J Physiol Endocrinol Metab. 2009;297(1):E260-9.
- Deevska G, Sunkara M, Karakashian C, Peppers B, Morris AJ, Nikolova-Karakashian MN. Effect of procysteine on aging-associated changes in hepatic GSH and SMase: evidence for transcriptional regulation of smpd3. J Lipid Res. 2014;55(10):2041-52.
- Bloomer SA, Han O, Kregel KC, Brown KE. Altered expression of iron regulatory proteins with aging is associated with transient hepatic iron accumulation after environmental heat stress. Blood Cells Mol Dis. 2014;52(1):19-26.
- Franko A, von Kleist-Retzow JC, Neschen S, Wu M, Schommers P, Bose M, et al. Liver adapts mitochondrial function to insulin resistant and diabetic states in mice. J Hepatol. 2014;60(4):816-23.
- Escande C, Nin V, Pirtskhalava T, Chini CC, Tchkonia T, Kirkland JL, et al. Deleted in breast cancer 1 limits adipose tissue fat accumulation and plays a key role in the development of metabolic syndrome phenotype. Diabetes. 2015;64(1):12-22.
- Di Naso FC, Porto RR, Fillmann HS, Maggioni L, Padoin AV, Ramos RJ, et al. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity (Silver Spring). 2015;23(1):120-9.
- Sanabria JR, Kombu RS, Zhang GF, Sandlers Y, Ai J, Ibarra RA, et al. Glutathione species and metabolomic prints in subjects with liver disease as biological markers for the detection of hepatocellular carcinoma. HPB (Oxford). 2016;18(12):979-90.
- Ghiraldini FG, Silveira AB, Kleinjan DA, Gilbert N, Mello ML. Genomic profiling of type-1 adult diabetic and aged normoglycemic mouse liver. BMC Endocr Disord. 2014;14:19.
- D'Souza AM, Asadi A, Johnson JD, Covey SD, Kieffer TJ. Leptin deficiency in rats results in hyperinsulinemia and impaired glucose homeostasis. Endocrinology. 2014;155(4):1268-79.
- Bianco A, Nigro E, Monaco ML, Matera MG, Scudiero O, Mazzarella G, et al. The burden of obesity in asthma and COPD: Role of adiponectin. Pulm Pharmacol Ther. 2017;43:20-5.
- Gairolla J, Kler R, Modi M, Khurana D. Leptin and adiponectin: pathophysiological role and possible therapeutic target of inflammation in ischemic stroke. Rev Neurosci. 2017;28(3):295-306.
- Perez-Diaz S, Johnson LA, DeKroon RM, Moreno-Navarrete JM, Alzate O, Fernandez-Real JM, et al. Polymerase I and transcript release factor (PTRF) regulates adipocyte differentiation and determines adipose tissue expandability. FASEB J. 2014;28(8):3769-79.
- Fok WC, Bokov A, Gelfond J, Yu Z, Zhang Y, Doderer M, et al. Combined treatment of rapamycin and dietary restriction has a larger effect on the transcriptome and metabolome of liver. Aging Cell. 2014;13(2):311-9.
- Fok WC, Chen Y, Bokov A, Zhang Y, Salmon AB, Diaz V, et al. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS One. 2014;9(1):e83988.
- Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol. 2015;33:55-66.
- Gunsar F. Liver Transplantation for Hepatocellular Carcinoma Beyond the Milan Criteria. Exp Clin Transplant. 2017;15(Suppl 2):59-64.
- Lee HW, Suh KS. Advancements of liver transplantation for hepatocellular carcinoma in Korea. Jpn J Clin Oncol. 2017;47(2):93-100.
- Cabral M, Martin-Venegas R, Moreno JJ. Differential cell growth/apoptosis behavior of 13-hydroxyoctadecadienoic acid enantiomers in a colorectal cancer cell line. Am J Physiol Gastrointest Liver Physiol. 2014;307(6):G664-71.
- Chen H, Cai W, Chu ES, Tang J, Wong CC, Wong SH, et al. Hepatic cyclooxygenase-2 over expression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene. 2017;36(31): 4415-26.
- Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, et al. Tissue regeneration. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science. 2015;348(6240):aaa2340.
- Bellavista E, Martucci M, Vasuri F, Santoro A, Mishto M, Kloss A, et al. Lifelong maintenance of composition, function and cellular/subcellular distribution of proteasomes in human liver. Mech Ageing Dev. 2014;141-142:26-34.
- Hodgson R, Christophi C. What determines ageing of the transplanted liver? HPB (Oxford). 2015;17(3):222-5.
- Fu H, Xu H, Chen H, Li Y, Li W, Zhu Q, et al. Inhibition of glycogen synthase kinase 3 ameliorates liver ischemia/reperfusion injury via an energy-dependent mitochondrial mechanism. J Hepatol. 2014;61(4):816-24.
- Cywes R, Greig PD, Morgan GR, Sanabria JR, Clavien PA, Harvey PR, et al. Rapid donor liver nutritional enhancement in a large animal model. Hepatology. 1992;16(5):1271-9.
- Cywes R, Greig PD, Sanabria JR, Clavien PA, Levy GA, Harvey PR, et al. Effect of intraportal glucose infusion on hepatic glycogen content and degradation, and outcome of liver transplantation. Ann Surg. 1992;216(3):235-46.
- Bellomo R, Marino B, Starkey G, Fink M, Wang BZ, Eastwood GM, et al. Extended normothermic extracorporeal perfusion of isolated human liver after warm ischaemia: a preliminary report. Crit Care Resusc. 2014;16(3):197-201.
- Bral M, Gala-Lopez B, Bigam D, Kneteman N, Malcolm A, Livingstone S, et al. Preliminary Single-Center Canadian Experience of Human Normothermic Ex Vivo Liver Perfusion: Results of a Clinical Trial. Am J Transplant. 2017;17(4):1071-80.
- Goldaracena N, Barbas AS, Selzner M. Normothermic and subnormothermic ex-vivo liver perfusion in liver transplantation. Curr Opin Organ Transplant. 2016;21(3):315-21.
- Ikeda T, Yanaga K, Lebeau G, Higashi H, Kakizoe S, Starzl TE. Hemodynamic and biochemical changes during normothermic and hypothermic sanguinous perfusion of the porcine hepatic graft. Transplantation. 1990;50(4):564-7.
- Nassar A, Liu Q, Farias K, D'Amico G, Tom C, Grady P, et al. Ex vivo normothermic machine perfusion is safe, simple, and reliable: results from a large animal model. Surg Innov. 2015;22(1):61-9.
- Ravikumar R, Jassem W, Mergental H, Heaton N, Mirza D, Perera MT, et al. Liver Transplantation After Ex Vivo Normothermic Machine Preservation: A Phase 1 (First-in-Man) Clinical Trial. Am J Transplant. 2016;16(6):1779-87.
- Selzner M, Goldaracena N, Echeverri J, Kaths JM, Linares I, Selzner N, et al. Normothermic ex vivo liver perfusion using steen solution as perfusate for human liver transplantation: First North American results. Liver Transpl. 2016;22(11):1501-8.
- Vogel T, Brockmann JG, Friend PJ. Ex-vivo normothermic liver perfusion: an update. Curr Opin Organ Transplant. 2010;15(2):167-72.
- Aravinthan A, Challis B, Shannon N, Hoare M, Heaney J, Alexander GJ. Selective insulin resistance in hepatocyte senescence. Exp Cell Res. 2015;331(1):38-45.
- Abbas R, Kombu RS, Ibarra RA, Goyal KK, Brunengraber H, Sanabria JR. The dynamics of glutathione species and ophthalmate concentrations in plasma from the VX2 rabbit model of secondary liver tumors. HPB Surg. 2011;2011:709052.
- Andres IR, Abbas R, Kombu RS, Zhang GF, Jacobs G, Lee Z, et al. Disturbances in the glutathione/ophthalmate redox buffer system in the woodchuck model of hepatitis virus-induced hepatocellular carcinoma. HPB Surg. 2011;2011:789323.
- Carulli L, Anzivino C. Telomere and telomerase in chronic liver disease and hepatocarcinoma. World J Gastroenterol. 2014;20(20):6287-92.
- Dlouha D, Maluskova J, Kralova Lesna I, Lanska V, Hubacek JA. Comparison of the relative telomere length measured in leukocytes and eleven different human tissues. Physiol Res. 2014;63 Suppl 3:S343-50.
- Kiran S, Oddi V, Ramakrishna G. Sirtuin 7 promotes cellular survival following genomic stress by attenuation of DNA damage, SAPK activation and p53 response. Exp Cell Res. 2015;331(1):123-41.
- uenantin AC, Briand N, Bidault G, Afonso P, Bereziat V, Vatier C, et al. Nuclear envelope-related lipodystrophies. Semin Cell Dev Biol. 2014;29:148-57.
- Arriazu E, Ruiz de Galarreta M, Cubero FJ, Varela-Rey M, Perez de Obanos MP, Leung TM, et al. Extracellular matrix and liver disease. Antioxid Redox Signal. 2014;21(7):1078-97.
- Irvine KM, Skoien R, Bokil NJ, Melino M, Thomas GP, Loo D, et al. Senescent human hepatocytes express a unique secretory phenotype and promote macrophage migration. World J Gastroenterol. 2014;20(47):17851-62.
- Idrissova L, Malhi H, Werneburg NW, LeBrasseur NK, Bronk SF, Fingas C, et al. TRAIL receptor deletion in mice suppresses the inflammation of nutrient excess. J Hepatol. 2015;62(5):1156-63.
- Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014;34(6):834-43.
- Abbas R, Adam SJ, Okadal S, Groar H, Anderson J, Sanabria J. Development of a swine model of secondary liver tumor from a genetically induced swine fibroblast cell line. HPB (Oxford). 2008;10(3):204-10.
- Ibarra R, Dazard JE, Sandlers Y, Rehman F, Abbas R, Kombu R, et al. Metabolomic Analysis of Liver Tissue from the VX2 Rabbit Model of Secondary Liver Tumors. HPB Surg. 2014;2014:310372.
- Hong X, Song R, Song H, Zheng T, Wang J, Liang Y, et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut. 2014;63(10):1635-47.
- Hong IH, Lewis K, Iakova P, Jin J, Sullivan E, Jawanmardi N, et al. Age-associated change of C/EBP family proteins causes severe liver injury and acceleration of liver proliferation after CCl4 treatments. J Biol Chem. 2014;289(2):1106-18.
- Hofmann JW, McBryan T, Adams PD, Sedivy JM. The effects of aging on the expression of Wnt pathway genes in mouse tissues. Age (Dordr). 2014;36(3):9618.
- Lu WJ, Chua MS, So SK. Suppressing N-Myc downstream regulated gene 1 reactivates senescence signaling and inhibits tumor growth in hepatocellular carcinoma. Carcinogenesis. 2014;35(4):915-22.
- Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. 2014;2:4172.
- Lopez-Dominguez JA, Khraiwesh H, Gonzalez-Reyes JA, Lopez-Lluch G, Navas P, Ramsey JJ, et al. Dietary fat and aging modulate apoptotic signaling in liver of calorie-restricted mice. J Gerontol A Biol Sci Med Sci. 2015;70(4):399-409.
- Jin J, Hong IH, Lewis K, Iakova P, Breaux M, Jiang Y, et al. Cooperation of C/EBP family proteins and chromatin remodeling proteins is essential for termination of liver regeneration. Hepatology. 2015;61(1):315-25.
- Sadri AR, Jeschke MG, Amini-Nik S. Advances in Liver Regeneration: Revisiting Hepatic Stem/Progenitor Cells and Their Origin. Stem Cells Int. 2016;2016:7920897.
- Sanabria JR, Gordon ER, Harvey PR, Goresky CA, Strasberg SM. Accumulation of un conjugated bilirubin in cholesterol pellets implanted in swine gallbladders. Gastroenterology. 1996;110(2):607-13.
- Sanabria JR, Upadhya A, Mullen B, Harvey PR, Strasberg SM. Effect of deoxycholate on immunoglobulin G concentration in bile: studies in humans and pigs. Hepatology. 1995;21(1):215-22.
- Ahsan MK, Mehal WZ. Activation of adenosine receptor A2A increases HSC proliferation and inhibits death and senescence by down-regulation of p53 and Rb. Front Pharmacol. 2014;5:69.
- Borkham-Kamphorst E, Schaffrath C, Van de Leur E, Haas U, Tihaa L, Meurer SK, et al. The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-beta signaling. Biochim Biophys Acta. 2014;1843(5):902-14.
- Ahluwalia A, Jones MK, Szabo S, Tarnawski AS. Aging impairs transcriptional regulation of vascular endothelial growth factor in human microvascular endothelial cells: implications for angiogenesis and cell survival. J Physiol Pharmacol. 2014;65(2):209-15.
- Ahluwalia A, Jones MK, Tarnawski AS. Key role of endothelial importin-alpha in VEGF expression and gastric angiogenesis: novel insight into aging gastropathy. Am J Physiol Gastrointest Liver Physiol. 2014;306(4):G338-45.
- Coletta C, Modis K, Olah G, Brunyanszki A, Herzig DS, Sherwood ER, et al. Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: preclinical studies in a murine model of cecal ligation and puncture. Crit Care. 2014;18(5):511.
- Hayashi T, Kotani H, Yamaguchi T, Taguchi K, Iida M, Ina K, et al. Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc Natl Acad Sci U S A. 2014;111(3):1168-73.
- Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 2017;66(1):212-27.
- Donato AJ, Henson GD, Hart CR, Layec G, Trinity JD, Bramwell RC, et al. The impact of ageing on adipose structure, function and vasculature in the B6D2F1 mouse: evidence of significant multisystem dysfunction. J Physiol. 2014;592(18):4083-96.
- Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez R-M, Shivakumar P, et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatology Communications. 2017;1(4):299-310.
- Ehlken H, Krishna-Subramanian S, Ochoa-Callejero L, Kondylis V, Nadi NE, Straub BK, et al. Death receptor-independent FADD signaling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific NEMO knockout. Cell Death Differ. 2014;21(11):1721-32.
- Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531(7593):253-7.
- Khraiwesh H, Lopez-Dominguez JA, Fernandez del Rio L, Gutierrez-Casado E, Lopez-Lluch G, Navas P, et al. Mitochondrial ultrastructure and markers of dynamics in hepatocytes from aged, calorie restricted mice fed with different dietary fats. Exp Gerontol. 2014;56:77-88.
- Girardet C, Begriche K, Ptitsyn A, Koza RA, Butler AA. Unravelling the mysterious roles of melanocortin-3 receptors in metabolic homeostasis and obesity using mouse genetics. Int J Obes Suppl. 2014;4(Suppl 1):S37-44.
- Ahn M, Park JS, Chae S, Kim S, Moon C, Hyun JW, et al. Hepatoprotective effects of Lycium chinense Miller fruit and its constituent betaine in CCl4-induced hepatic damage in rats. Acta Histochem. 2014;116(6):1104-12.
- Bae KH, Min AK, Kim JG, Lee IK, Park KG. Alpha lipoic acid induces hepatic fibroblast growth factor 21 expression via up-regulation of CREBH. Biochem Biophys Res Commun. 2014;455(3-4):212-7.
- Ip BC, Liu C, Ausman LM, von Lintig J, Wang XD. Lycopene attenuated hepatic tumorigenesis via differential mechanisms depending on carotenoid cleavage enzyme in mice. Cancer Prev Res (Phila). 2014;7(12):1219-27.
- Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X, et al. Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK-dependent autophagy. Int J Biol Sci. 2014;10(3):268-77.
- Momchilova A, Petkova D, Staneva G, Markovska T, Pankov R, Skrobanska R, et al. Resveratrol alters the lipid composition, metabolism and peroxide level in senescent rat hepatocytes. Chem Biol Interact. 2014;207:74-80.
- Park KH, Kim JM, Cho KH. Elaidic acid (EA) generates dysfunctional high-density lipoproteins and consumption of EA exacerbates hyperlipidemia and fatty liver change in zebrafish. Mol Nutr Food Res. 2014;58(7):1537-45.
- Spartano NL, Lamon-Fava S, Matthan NR, Obin MS, Greenberg AS, Lichtenstein AH. Linoleic acid suppresses cholesterol efflux and ATP-binding cassette transporters in murine bone marrow-derived macrophages. Lipids. 2014;49(5):415-22.
- Yao WL, Ko BS, Liu TA, Liang SM, Liu CC, Lu YJ, et al. Cordycepin suppresses integrin/FAK signaling and epithelial-mesenchymal transition in hepatocellular carcinoma. Anticancer Agents Med Chem. 2014;14(1):29-34.
- Zhang J, Wang M, Zhang Z, Luo Z, Liu F, Liu J. Celecoxib derivative OSU-03012 inhibits the proliferation and activation of hepatic stellate cells by inducing cell senescence. Mol Med Rep. 2015;11(4):3021-6.
- Hu B, Du Q, Deng S, An HM, Pan CF, Shen KP, et al. Ligustrum lucidum Ait. fruit extract induces apoptosis and cell senescence in human hepatocellular carcinoma cells through upregulation of p21. Oncol Rep. 2014;32(3):1037-42.
- Hasan ST, Zingg JM, Kwan P, Noble T, Smith D, Meydani M. Curcumin modulation of high fat diet-induced atherosclerosis and steatohepatosis in LDL receptor deficient mice. Atherosclerosis. 2014;232(1):40-51.
- Drummond CA, Hill MC, Shi H, Fan X, Xie JX, Haller ST, et al. Na/K-ATPase signaling regulates collagen synthesis through microRNA-29b-3p in cardiac fibroblasts. Physiol Genomics. 2016;48(3):220-9.
- Hangaard L, Bouzinova EV, Staehr C, Dam VS, Kim S, Xie Z, et al. Na-K-ATPase regulates intercellular communication in the vascular wall via cSrc kinase-dependent connexin43 phosphorylation. Am J Physiol Cell Physiol. 2017;312(4):C385-C97.
- Li Z, Cai T, Tian J, Xie JX, Zhao X, Liu L, et al. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biol Chem. 2009;284(31):21066-76.
- Li Z, Zhang Z, Xie JX, Li X, Tian J, Cai T, et al. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J Biol Chem. 2011;286(37):32394-403.
- Liu J, Tian J, Chaudhry M, Maxwell K, Yan Y, Wang X, et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Sci Rep. 2016;6:34592.
- Sodhi K, Maxwell K, Yan Y, Liu J, Chaudhry MA, Getty M, et al. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci Adv. 2015;1(9):e1500781.
- Sodhi K, Srikanthan K, Goguet-Rubio P, Nichols A, Mallick A, Nawab A, et al. pNaKtide Attenuates Steatohepatitis and Atherosclerosis by Blocking Na/K-ATPase/ROS Amplification in C57Bl6 and ApoE Knockout Mice Fed a Western Diet. Sci Rep. 2017;7(1):193.
- Srikanthan K, Shapiro JI, Sodhi K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Obesity and Cardiovascular Disease. Molecules. 2016;21(9).
- Wang Y, Ye Q, Liu C, Xie JX, Yan Y, Lai F, et al. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic Biol Med. 2014;71:415-26.